基质相互作用分子1 (STIM1)参与介导储存操作的Ca2+进入(SOCE),驱动细胞内第二信使钙离子(Ca2+)的内流,与肿瘤细胞的增殖、转移、凋亡、自噬、代谢和免疫过程密切相关。STIM1不仅在转录水平上受NF-κB和HIF-1的调控,还可被mirna转录后修饰并被泛素化降解。最近的研究表明,STIM1或Ca2+信号可以调节肿瘤细胞的凋亡、自噬、焦亡和铁亡,并且在不同的癌症中作用不同。此外,STIM1通过影响肿瘤细胞死亡,有助于抵抗抗肿瘤治疗。进一步研究STIM1控制其他形式肿瘤细胞死亡的机制有助于发现新的治疗靶点。此外,STIM1具有调节肿瘤微环境内免疫细胞的能力。本文综述了STIM1的基本结构、功能和调控,总结了STIM1调控肿瘤细胞死亡的信号通路,并提出了以STIM1为靶点的抗肿瘤治疗前景。
STIM1是构成CRAC的主要分子之一,CRAC是驱动Ca2+内流实现SOCE功能的通道,与肿瘤细胞增殖、转移、死亡和免疫密切相关。
STIM1可以通过PI3K-SGK1-P300-NF-κB和CaMKKII-P300-HIF-1信号通路在转录水平上进行调控,也可以通过各种mirna进行转录后修饰,也可以通过泛素化直接降解。
凋亡、自噬、焦亡、铁亡和铜腐是程序性细胞死亡的典型形式,而STIM1能够影响肿瘤细胞中这些细胞死亡的发生。
STIM1通过增强免疫细胞的增殖、激活和记忆来增强肿瘤细胞死亡和抗肿瘤免疫,从而增强抗肿瘤治疗。
发现靶向STIM1促进肿瘤细胞死亡的有效新药是未来抗肿瘤治疗的一个有希望的方向。
在不同类型的癌症中,STIM1影响肿瘤细胞死亡的各种途径是什么?
STIM1如何通过影响细胞死亡来调节抗肿瘤治疗的耐药性和肿瘤免疫?
靶向STIM1治疗人类肿瘤的潜在应用是什么?
钙离子(Ca2+)是细胞内重要的第二信使。细胞内Ca2+浓度的变化控制着细胞增殖、凋亡、代谢和免疫等多种细胞活动[1,2,3,4]。在不可兴奋的细胞中,如肿瘤细胞,细胞外Ca2+通过质膜(PM)内流的主要途径是Ca2+释放激活的Ca2+通道(CRAC),其介导储存操作的Ca2+进入(SOCE)。SOCE的功能已被广泛研究,但其调控机制一直是一个谜,直到2005年[5]和2006年[6],基质相互作用分子1 (STIM1)和Orai1被确定为维持SOCE功能的关键分子。STIM1是一种跨越内质网(ER)膜的跨膜蛋白,其n端锚定在ER膜上,c端位于细胞质中[7],而Orai1是一种位于PM上的蛋白,对Ca2+具有高度选择性[8]。当内质网的Ca2+池被耗尽时,STIM1感知到信号并寡聚,随后STIM1易位到PM,激活ER-PM连接处的Orai1,从而激活CRAC和SOCE,引起Ca2+内流[9]。除了激活Orai1外,STIM1还可以激活哺乳动物体内的Orai2和Orai3产生ICRAC,但效率较低[10]。值得注意的是,标准TRP (TRPC)通道以及STIM1和Orai1都参与了SOCE[11]。Guido等人证明,即使没有STIM1蛋白,位于内质网中的单一跨膜蛋白junctate也可以通过募集内质网连接来介导SOCE[12]。这些发现表明,SOCE可以在不特异性靶向STIM1蛋白的情况下被触发,为癌症治疗提供了一种新的方法。
最近,人们越来越清楚地发现STIM1参与调节多种癌症的发生、发展、侵袭和转移。在这些过程中,肿瘤细胞死亡起着至关重要的作用。根据是否受基因调控,细胞死亡可分为意外细胞死亡(ACD)和调节细胞死亡(RCD)两种[13,14,15]。最近有令人信服的证据表明,诱导肿瘤细胞死亡可以增强抗肿瘤免疫治疗的效果[16,17,18],而stim1诱导的肿瘤细胞死亡可以减轻抗肿瘤治疗的耐药性[19,20,21]。
在此背景下,我们综述了STIM1的发现、结构、激活过程、调控STIM1的上游信号通路,总结了STIM1在肿瘤细胞死亡中的功能以及通过影响肿瘤细胞死亡逆转抗肿瘤治疗耐药的途径,以及STIM1在肿瘤免疫中的关键作用。最后,我们讨论了STIM1的临床应用及未来展望。
2005年,Parekh AB和Putney JW Jr提出细胞膜中存在四种主要类型的Ca2+通道,存储操作通道(SOC)被认为是Ca2+进入细胞的最原始途径[22]。虽然STIM最早由Parker于1996年发现[23],但其功能并不明确。Roos通过RNA干扰(RNAi)抑制果蝇S2细胞中的多个基因,发现STIM1是构成thapsigargin (TG)依赖性Ca2+内流和CRAC通道的分子基础[5]。
STIM编码一种二聚体I型跨膜蛋白,在哺乳动物中有两个同源物STIM1和STIM2。STIM1位于人类染色体11p15.5区域[23,24]。STIM1由三个不同的组成部分组成:位于内质网(ER)管腔的n端、细胞质链和可溶于细胞质溶胶的c端[25]。n端部分包括典型EF-hand (cEF)、隐藏EF-hand (hEF)和不育α基序(SAM)。细胞质链区由三个被称为CC1、CC2和CC3的螺旋状结构域组成。CC1和SAM结构域通过跨膜结构域(TM)连接。此外,stim - orai激活区(SOAR)也被称为crac激活域(CAD),由来自CC2和CC3的片段组成。此外,c端段由Pro/Ser-rich区和Lys-rich区组成[7,26](图1A)。在过去的十年中,通过核磁共振波谱和x射线晶体学已经更好地了解了STIM1结构域及其在激活过程中的构象变化[26,27,28,29]。
图1:STIM1的位置和结构。

STIM1主要定位于内质膜,在PM中有一小部分。Orai1是一种质膜蛋白,具有四个跨膜区和细胞内N端和c端。当内质网腔内的Ca2+被耗尽时,EF-SAM复合物分解。随后,STIM1寡聚并易位到PM上,然后与PM上的Orai1结合,激活CRAC并引起Ca2+内流。B预测STIM1的结构域。从n端开始,STIM1包括一个典型的EF-hand (nEF),一个隐藏的EF-hand (hEF)和管腔侧的无菌基序(SAM)结构域。细胞质一侧是重叠的螺旋状结构域(CC)和富含丝氨酸-脯氨酸(S/P)和赖氨酸(K)的结构域。
STIM1有两个主要功能:首先,它能够感知内质腔内Ca2+ (100-400 μM)浓度的精确变化[30],其次,它将该信号传递给PM上的Orai1并激活SOCE。
当细胞处于静止状态时,STIM1的cEF-hand负责与内质网腔Ca2+结合,hEF-hand维持稳定性[31]。当PM上的G蛋白偶联受体(gpcr)或蛋白酪氨酸激酶连接受体(PTKRs)被激素和生长因子激活时,它们与相应的磷脂酶C (PLC)异构体结合,分别产生1,4,5-三磷酸(InsP3)和二酰基甘油(DAG)。随后,InsP3与ER膜上的InsP3R结合,导致Ca2+释放[2]。当内质网腔内Ca2+浓度低于300 μM (Hill系数约为4)时,它与STIM1的cEF-hand分离,从而使EF-SAM复合物不稳定,导致STIM1寡聚化[32]。紧接着,STIM1转运到ER-PM连接位点,并与Orai1发生物理相互作用,打开CRAC通道,导致Ca2+进入[30,33]。此外,可靠的实验表明,STIM1的细胞质c端是打开Orai1通道的关键结构域[34](图1B)。通过诱导细胞突变和亚细胞定位,STIM1在构建CRAC通道和激活SOCE中的关键功能已逐渐被阐明。
迄今为止,已经鉴定和研究了STIM1的三种剪接变异亚型。第一个亚型STIM1L于2011年被发现,在小鼠的大脑和心脏、新生大鼠的心肌细胞和人类的骨骼肌细胞中特异性表达。STIM1L是通过外显子11的选择性剪接产生的。它的c端区域与另外106个残基一起形成永久性的肌动蛋白结合域(ABD),导致SOCE激活的速率和频率显著增加[35]。第二种亚型,称为STIM1A或STIM1β,于2020年被发现,在星形胶质细胞、心脏、肾脏和睾丸中高度表达。它是通过在STIM1的c端491位插入额外的31个氨基酸残基而形成的[36]。Xie等人发现,在胶质母细胞瘤中上调的STIM1β可以作为Orai1通道的有效激活剂,从而增强SOCE的功能[37]。另一方面,全长STIM1A负调控SOCE和ICRAC,但它通过减少cAMP降解来增强NFAT易位[38]。另一种变体STIM1B于2021年被发现,在中枢神经系统(CNS)中含量很高。STIM1B是通过在STIM1的细胞质区域插入一个特定的基序而产生的,这使得它能够被特异性靶向。实验结果表明,STIM1B向ER-PM连接处缓慢募集,导致SOCE的速率和峰值降低,以及ICRAC的减少[39]。
摘要。
事实。
开放式的问题
介绍
STIM1:发现、结构和激活
STIM1的上游调控途径
STIM1在肿瘤细胞死亡中的作用
STIM1介导抗肿瘤治疗耐药性
STIM1在肿瘤免疫中的作用
靶向STIM1的治疗潜力
有限公司
结论和观点
数据可用性
参考文献。
致谢。
作者信息
道德声明
# # # # #
STIM1/Orai是CRAC通道的基本组成部分,在SOCE介导的细胞内Ca2+稳态调节中起重要作用。各种细胞内信号通过调节STIM1/Orai影响细胞内Ca2+浓度(图2)。
图2:STIM1的上游调控途径。
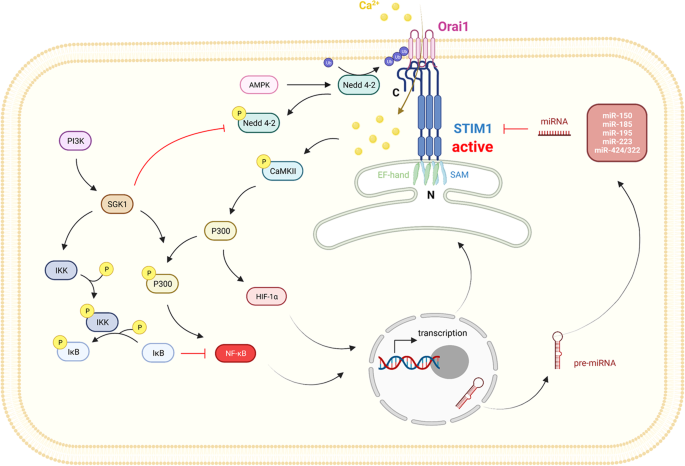
STIM1和Orai1的转录水平可通过多种信号通路调节,包括PI3K-SGK1-NF-κB和CaMKII-P300-HIF-1α轴。此外,AMPK-Nedd4-2可触发Orai1的泛素化。在转录后修饰水平上,多种肿瘤中STIM1的表达水平受到miR-150、miR-185、miR-195、miR-223和miR-424/322等多种mirna的抑制。
例如,转录因子核因子κB (NF-κB)已被证明可以增加STIM1和Orai1的mRNA和蛋白水平[40,41,42]。进一步的实验表明,NF-κB通过p65分别结合STIM1和Orai1启动子的S1和O2区域来促进它们的转录。此外,研究表明NF-κB的p65/p50或p65/p52亚基通过转录激活STIM1和Orai1来增强HEK293细胞的迁移能力[43]。我们早期的研究报道了缺氧诱导因子-1 (HIF-1)可以转录STIM1以增强肝细胞癌(HCC)细胞的SOCE[44]。
除了调节STIM1和Orai1的转录水平外,Keil发现大量的SH3 's (POSH),一种E3泛素连接酶,可以直接泛素化STIM1并启动其降解,从而减少细胞内Ca2+[45]。同样,研究表明,神经元前体细胞表达发育下调的4-2 (Nedd4-2),也是E3泛素连接酶,能够泛素化并降解Orai1,导致Orai1不能被STIM1激活以执行SOCE功能[46]。在调节Nedd4-2方面,Almaca等人发现AMPK可以激活它[47,48],后来Nurbaeva等人也报道了AMPK缺陷小鼠树突状细胞(dc)中STIM1和Orai1的高表达[49]。
血清和糖皮质激素诱导激酶1 (SGK1)是磷脂酰肌醇3-激酶(PI3K)信号传导的下游靶点[50,51,52]。实验表明,sgk1缺陷小鼠中STIM1和Orai1的转录水平明显低于对照组[53]。此外,SGK1可以磷酸化Nedd4-2,从而逆转Ca2+内流的减少[46]。总体而言,SGK1通过上调STIM1/Orai1促进细胞迁移、肥大细胞脱颗粒和血小板聚集,从而促进SOCE功能[54,55]。
MicroRNAs (miRNAs)参与基因表达的转录后调控[56]。最近,已经发现mirna通过转录后修饰STIM1 mRNA的3 ' -非翻译区(3 ' -UTR)来调节各种组织中的STIM1。一方面,在正常细胞中,如肠上皮细胞中,miR-195与rna结合蛋白HUR竞争,结合STIM1 mRNA的3'UTR,使其不稳定,从而抑制细胞迁移[57]。此外,在血管平滑肌细胞(VSMC)中,miR-424/322通过损害STIM1和cyclin D1来阻止VSMC增殖和组织修复[58]。另一方面,在肿瘤细胞中,如结直肠癌(CRC)细胞中,miR-185可以钝化STIM1的表达,从而抑制CRC的转移和侵袭[59]。此外,在腔内非侵袭性MCF7乳腺癌细胞中,miR-223和miR-150可以降低STIM1的表达,但这种调节对乳腺癌的影响尚不清楚[60]。
综上所述,在不同器官的肿瘤发生、进展和转移过程中,STIM1和Orai1的转录、转录后修饰和翻译水平受到多种信号通路的调节。此外,STIM1和Orai1可以被泛素化直接降解。靶向这些特异性信号分子是一种潜在的抗肿瘤治疗策略。
自STIM1被发现以来,其在血小板聚集[52]、过敏反应[61]、自身免疫性疾病[62]、肿瘤细胞死亡[63]和t细胞分化[64]中的作用不断被研究。肿瘤是一种以细胞增殖和死亡失衡为特征的疾病。细胞在特定刺激下以不同的模式死亡,细胞死亡信号通路的获得性缺陷是肿瘤的一个特征。各种抗肿瘤治疗的目的都是干扰和阻断肿瘤细胞的增殖和转移过程,从而诱导肿瘤细胞死亡。因此,弄清肿瘤细胞死亡的机制是优化抗肿瘤治疗措施的关键。本文总结了STIM1调控的肿瘤细胞死亡及其机制(表1)。
表1 STIM1在肿瘤细胞死亡中的作用。
细胞凋亡是一种PCD,其基本功能是维持组织稳态[65]。癌症被认为是一种细胞过度增殖的疾病,促进肿瘤细胞凋亡可有效抑制肿瘤进展[66]。Ca2+已被证明可调节多种肿瘤的细胞凋亡,但其机制尚不明确[67]。
在大多数类型的肿瘤中,STIM1减弱细胞凋亡。例如,在胰腺癌中,STIM1和Orai1对细胞凋亡有抑制作用。5-氟尿嘧啶(5-FU)和吉西他滨等化疗药物可通过阻断STIM1和Orai1增强胰腺癌细胞凋亡[19]。在胃癌中,下调STIM1也可促进细胞凋亡[68]。此外,Chakraborty的研究表明,在三阴性乳腺癌(TNBC)细胞中,phenindoole可延迟STIM1向PM的易位,从而减少其与Orai1的结合,引起内质网应激并诱导细胞凋亡[69]。STIM1不仅在乳腺癌中高表达,而且在89%的头颈部鳞状细胞癌(HNSCC)中表达增加。Li的研究表明,在HNSCC中通过抑制STIM1表达来降低细胞内Ca2+浓度可以加速内质网应激相关的细胞死亡,如凋亡[70]。
然而,在前列腺癌(PCa)中,STIM1起相反的作用。Flourakis发现orai1诱导的Ca2+进入是导致PCa细胞凋亡的主要因素。在PCa细胞LNCaP中转染突变体R91W和L273S,破坏Orai1功能,可降低SOCE,从而保护LNCaP细胞免受tg诱导的凋亡[71]。
综上所述,靶向肿瘤细胞凋亡是一种很有前途的抗肿瘤治疗策略。然而,STIM1在不同肿瘤中的作用存在很大的异质性,这可能与它们不同的细胞类型和信号通路有关。因此,对于不同类型的肿瘤,需要对STIM1调控细胞凋亡的分子机制进行差异性研究。
自噬是将细胞质和细胞器分离到自噬体中,并将其转运到溶酶体进行蛋白水解的过程[72]。自噬的本质是维持细胞和机体的能量稳态[73]。近十年的研究表明,STIM1在诱导肿瘤细胞自噬中起着重要作用。
急性早幼粒细胞白血病(APL)是一种非实体肿瘤,全反式维甲酸(ATRA)是其治疗的一线选择[74]。既往研究表明,ATRA在APL患者粒细胞分化过程中诱导自噬,但上游机制尚不清楚[75]。Merhi证明ATRA通过SOCE/钙调素依赖性蛋白激酶激酶2 (CAMKK2)/AMPK信号轴激活自噬,这些分子可能是增强ATRA抗癌作用的潜在靶点[76](图3A)。
图3:STIM1通过SOCE调控AMPK信号通路诱导四种肿瘤细胞自噬的机制示意图。

在APL中,ATRA通过激活STIM1上调p-CaMKK2和P-AMPK,进而抑制mTOR表达,导致自噬。随后PML-RARα癌蛋白的降解使APL经历粒细胞分化,从而转化为成熟状态。B DIM在胃癌细胞中被用来触发自噬。其机制是DIM上调stim1介导的SOCE,随后上调p-AMPK的表达,进而通过CHOP信号诱导内质网应激。C在HCC中,线粒体分裂和细胞质Ca2+信号通路之间形成了一个正反馈回路。一方面,STIM1通过提高p-CaMKK2和p-AMPK的表达诱导自噬;另一方面,通过NFATc2和c-Myc对Drp1和FIS1的转录激活,STIM1通过线粒体分裂促进ROS的产生,进而上调STIM1。D RSV具有促进PCa自噬的能力,与其他肿瘤不同,它依赖于阻断STIM1的表达,从而激活p-AMPK,抑制mTOR通路。
STIM1还可以通过AMPK调控实体肿瘤如胃癌的自噬。Ye发现3,3′-二吲哚基甲烷(DIM)通过上调stim1介导的SOCE增强p- ampk - chop介导的内质膜应激,进而促进胃癌细胞的自噬[77](图3B)。此外,Huang等人证明,在HCC细胞中,活性氧(ROS)增加激活的NF-κB通路可以上调STIM1,从而激活SOCE功能。细胞内Ca2+的增加进一步通过活化t细胞核因子2 (NFATc2)和c-myc促进动力蛋白相关蛋白1 (Drp1)和线粒体裂变1 (FIS1)的表达,从而引起线粒体裂变[78]。此外,细胞内Ca2+的增加也会刺激CaMKK/AMPK通路,从而导致HCC的自噬[79](图3C)。相反,Selvaraj的实验表明,白藜芦醇(RSV)通过抑制STIM1激活AMPK,进而下调V-akt小鼠胸腺瘤病毒癌基因同源物(AKT)/哺乳动物雷帕霉素靶点(mTOR)信号轴,最终促进PCa细胞的自噬[80]。此外,Kondratskyi的研究针对AKT-mTOR信号通路,阐明了STIM1和AKT的抑制剂ML-9可以通过下调mTOR的表达来促进PCa细胞的自噬[81](图3D)。此外,ML-9具有增强多西紫杉醇抗癌活性的作用,但其分子机制有待进一步研究。
综上所述,stim1 - ssoe - ampk自噬通路可能是血液学癌症和实体肿瘤的潜在治疗靶点。
目前对STIM1调控肿瘤细胞凋亡和自噬的机制研究较多,而STIM1与其他类型肿瘤细胞死亡(如焦亡、铁亡、铜亡)的关系尚不清楚。最近的研究表明,钙信号可以改变肿瘤细胞中这些PCDs的水平,但STIM1是否参与其中还需要进一步研究。
Pyroptosis
焦亡是caspase-1介导的单核细胞死亡,其特征是炎症小体的产生,caspase-11/4/5也在这一过程中被激活[82]。目前,被广泛研究的炎性小体传感器有节点样受体1 (NLRP1)、NLRP3和节点样核子4 (NLRC4)。
葫芦素B (Cucurbitacin B, CuB)可诱导肿瘤细胞凋亡,并对炎症反应有抑制作用[83,84]。Yuan等人发现,CuB与toll样受体4 (TLR4)结合,通过激活NLRP3炎性小体复合体,促进线粒体ROS的产生和细胞内Ca2+的积累,从而促进非小细胞肺癌(NSCLC)的焦亡[85]。硒蛋白具有重要的抗氧化和抗炎作用,最近的研究表明,摄入一定量的硒对某些肿瘤也有预防作用[86]。如Liu等研究表明,硒蛋白受体硫氧还蛋白还原酶3 (Txnrd3)在小鼠结肠中表达,导致内质网应激和Ca2+释放增加,进而促进NLRP3和Gasdermin D (GSDMD)的表达,促进焦亡,从而使结肠癌细胞的生长和增殖变得迟缓[87]。
Ferroptosis
Ferroptosis是Dixon于2012年发现的一种新型铁依赖性脂质活性氧(L-ROS)诱导的PCD[88]。
Chen等人首次发现,erianin可以抑制NSCLC细胞的增殖和迁移,并通过促进NSCLC中Ca/calmodulin (CaM)信号通路激活l型电压依赖性Ca2+通道(LVDCC),同时增加Ca2+转运和Fe2+水平,进一步促进L-ROS的产生,诱导铁凋亡[89]。线粒体对Ca2+信号传导和氧化还原稳态至关重要[90]。线粒体钙单转运蛋白(MCU)是调节线粒体内Ca2+稳态的重要复合物。MCU通过激活kelch样ech相关蛋白1- nf - e2相关因子2 (KEAP1-NRF2)抗氧化途径促进胱氨酸/谷氨酸反转运蛋白SLC7A11的表达,进而促进胰腺导管腺癌(pancreatic ductal adencarcinoma, PDAC)细胞的迁移、侵袭和代谢应激抵抗[91]。在黑色素瘤中也观察到nrf2依赖的抗氧化系统与铁下垂调节之间的关系。Wang等人发现,在黑色素瘤中,ampk介导的CAMKK2激活可促进NRF2表达,从而抑制l - ros依赖性铁下垂。靶向抑制AMPK-CAMKK2-NRF2信号轴可增强抗pd -1抗体在黑色素瘤中的疗效[92]。然而,STIM1是否以及如何调控铁下垂有待研究。
Cuproptosis
cuprotosis是Tsvetkov在2022年发现的最新PCD。其机制是铜与三羧酸(TCA)循环的脂酰化组分直接结合,导致脂酰化蛋白聚集,铁硫簇蛋白丢失,最终导致蛋白质毒性应激和细胞死亡[15]。Ca2+与Cu2+之间的密切联系已被发现[93,94],但Ca2+是否参与铜还原的调控还有待进一步研究。此外,它们在SOCE和铜凸之间的关系仍未被探索。
在过去的20年里,针对特定基因的靶向治疗已经在一些癌症中被提出,并且已经确定了调节癌症发展的分子机制,这已经彻底改变了抗肿瘤治疗策略。然而,尽管抗肿瘤治疗取得了重大进展,但不可避免的耐药性是改善患者预后的主要障碍。
逃避凋亡是肿瘤细胞的特征之一[95],也是导致治疗效率低下的重要原因。雄激素是早期前列腺癌的标准治疗方法,但不幸的是,前列腺癌会发展到以细胞凋亡抵抗为特征的不依赖雄激素的阶段[96]。Flourakis团队在2004年发现雄激素非依赖性PCa细胞的凋亡抵抗与SOCE下调有关[20]。随后,2010年,Flourakis证明了由Orai1下调引起的SOCE降低导致钙依赖性细胞凋亡受到抑制[76]。Dubois等人发现,PCa细胞中Orai3的过表达破坏了Orai1的同化,导致SOCE密度降低,引起细胞凋亡抵抗[97]。本文中,靶向STIM1/Orai1/SOCE分子有望逆转PCa细胞的凋亡抵抗。相比之下,在非实体肿瘤如B淋巴瘤中,沉默Orai1可增强利妥昔单抗的促凋亡作用。这表明利妥昔单抗联合CRAC通道抑制剂或Ca2+抑制剂可改善B淋巴瘤患者的预后[98]。
除了内分泌和靶向治疗外,肿瘤细胞对化疗的耐药性已被证明与STIM1抑制细胞凋亡有关。Sun等人发现,在顺铂耐药骨肉瘤MG63细胞中,STIM1显著上调。在MG63细胞中,敲低STIM1增加GRP78、CHOP和ATF4d的表达,诱导内质网应激,部分恢复顺铂耐药骨肉瘤细胞的敏感性[21]。5-FU也是抗肿瘤治疗中常用的化疗药物,具有诱导肿瘤细胞自噬的作用[99]。Tang的研究表明,5-FU通过阻断PI3K/AKT/mTOR信号轴导致HepG2细胞自噬。敲低Orai1或使用SOCE抑制剂SKF96365可进一步增强5-FU抑制这一途径的能力,从而使HepG2细胞对5-FU处理变得敏感[100]。因此,在B淋巴瘤、骨肉瘤和HCC等几种肿瘤中,STIM1可能保护它们避免逃避靶向治疗或化疗。在这些肿瘤中使用Ca2+或SOCE抑制剂可能部分恢复它们对抗肿瘤治疗的敏感性。
肿瘤由肿瘤实质细胞和肿瘤微环境组成。增强TME中免疫细胞的杀伤能力可引起肿瘤实质细胞的免疫原性细胞死亡(immunogenic cell death, ICD)[18,101]。同时,近年来的实验表明,Ca2+信号对淋巴细胞功能至关重要,stim1缺陷的哺乳动物会发生免疫缺陷[102,103,104]。
STIM1和Orai1在T细胞介导的免疫反应中上调,并在T细胞与抗原呈递dc接触的区域引起Ca2+内流增加。扩增的Ca2+信号对t细胞的激活、扩增和分化具有催化作用[105]。Weidinger发现STIM1通过增加CD8+ T细胞的FasL表达、穿孔素和颗粒酶B的分泌以及TNF-α和IFN-γ的释放来增强CD8+ T细胞的细胞毒作用[106](图4A)。此外,Shaw等人证明STIM1促进CD4+ T细胞表面CD40L的表达,从而促进CD8+ T细胞在再感染时的记忆反应[107](图4B)。此外,STIM1和calcalineurin (CaN)协同激活NFAT和PI3K-AKT-mTOR信号通路,通过促进葡萄糖摄取和利用来刺激na?ve t细胞增殖[108](图4C)。
图4:STIM1增强T细胞的肿瘤杀伤、记忆和增殖。
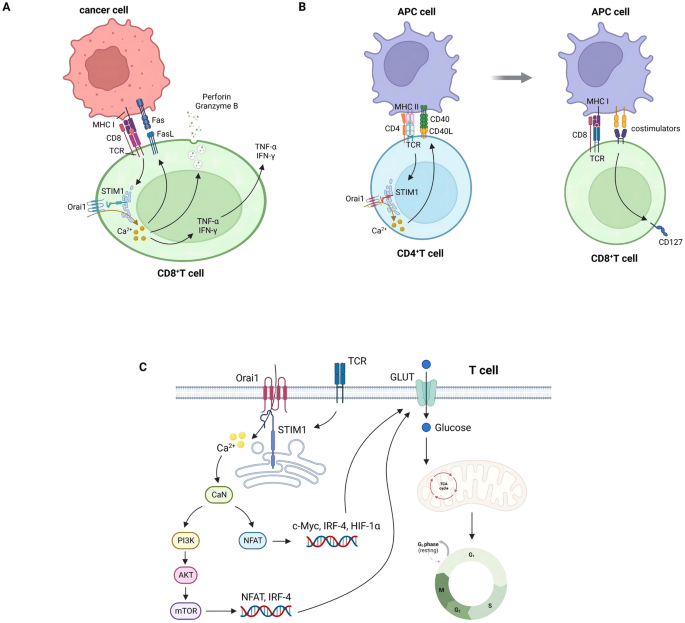
通过TCR刺激激活STIM1介导CD8+T细胞的脱颗粒、TNF-α和IFN-γ的产生以及FasL的表达,所有这些都是抗肿瘤免疫所必需的。B CD8+T细胞的记忆应答功能(通过CD127的表达)依赖于STIM1促进的CD4+T细胞中CD40L的表达,而CD40L的表达反过来又加速了apc的抗原呈递。C STIM1通过刺激CaN激活PI3K-AKT-mTOR和NFAT营养感应通路来调节t细胞增殖,从而提高葡萄糖的摄取和进入TCA的利用。
T细胞是适应性免疫的主要效应细胞,而中性粒细胞也是先天免疫应答的关键细胞。研究表明,在stim1缺失的小鼠中,中性粒细胞脱粒、吞噬和ROS的产生完全受损,这与蛋白激酶C亚型α和β (PKC-α和PKC-β)激活减少相关,导致氧化酶亚基磷酸化减少[109]。紧接着,Clemens等人发现STIM1和STIM2协同调节中性粒细胞中的SOCE功能。而STIM1是影响中性粒细胞杀菌功能的主要因子,而STIM2通过活化NF-κB调节中性粒细胞分泌TNF-α、IL-10和IFN-γ细胞因子[110]。
细胞毒性T细胞的激活依赖于dc的抗原呈递,从而激活它们对有害刺激的适应性免疫反应。Nunes-Hasler的研究表明STIM1对dc有选择性调控。在髓性stim1缺陷小鼠中,dc的吞噬作用和抗原交叉呈递减少。然而,小鼠髓源性dc中成熟标志物的上调并不需要STIM1,也不影响其分化[111]。总之,靶向STIM1增强T细胞对肿瘤细胞的细胞毒性作用是一种很有前景的治疗策略。
ccDownload: /内容/ pdf / 10.1038 / s41420 - 023 - 01703 - 8. - pdf