观察性研究表明,肠道微生物群与重症肌无力之间存在关联;然而,两者之间的因果关系仍不明确。我们的目标是确定肠道微生物群组成与重症肌无力之间存在双向因果关系,并研究肠道微生物群如何在降低重症肌无力风险中发挥作用。
我们从MiBioGen联盟(N=18,340)和FinnGen研究项目(426例和373,848例对照)中获得了门、类、目、科和属水平的肠道微生物群数据。在双样本孟德尔随机化分析中,我们评估了肠道微生物群与重症肌无力之间的因果关系。我们还进行了双向磁共振分析,以确定因果关系的方向。采用反方差加权、孟德尔随机化-艾格、加权中位数、简单模型和加权模型检验肠道菌群与重度重症肌无力的因果关系。我们分别使用MR-Egger截距和Cochran’s Q检验来评估多效性和异质性。此外,我们利用MR-PRESSO方法评估水平多效性并检测异常值。
在正向分析中,反方差加权法显示,Lachnoclostridium属与重症肌无力风险呈正相关(OR=2.431,95%CI 1.047 ~ 5.647, p=0.039)。此外,clostridiaceae科(OR=0.424,95%CI 0.202 ~ 0.889, p=0.023)、Defluviitaleaceae科(OR=0.537,95%CI 0.290 ~ 0.995, p=0.048)、Enterobacteriaceae科(OR=0.341,95%CI 0.135 ~ 0.865, p=0.023)和未知属(OR=0.407,95%CI 0.209 ~ 0.793, p=0.008)均与重症肌无力发生风险呈负相关。此外,反向孟德尔随机化分析证实重症肌无力与巴氏菌属的风险呈负相关(OR=0.945,95%CI 0.906 ~ 0.985, p=0.008)。
我们的研究证明了肠道微生物群和重症肌无力之间的双向因果关系。我们确定了与重症肌无力相关的特定类型的微生物,这为了解这种疾病的发病机制和开发治疗策略提供了新的窗口。然而,需要更多的基础和临床研究来阐明肠道微生物群在重症肌无力发病机制中的确切作用和治疗潜力。
重症肌无力(MG)是一种自身免疫性疾病,其特征是抗体攻击神经肌肉连接处,导致肌肉无力。最常见的症状是眼肌无力,也可累及球、肢体、轴肌和呼吸肌,进展为全身性MG[1]。临床上20%的MG患者出现需要机械通气的急性呼吸衰竭,导致病死率显著升高[2]。MG的病因是多方面的,包括遗传因素和多种环境危险因素[3,4,5]。然而,导致个体对MG易感性的确切因素尚不清楚。肠道菌群对人体肠道内宿主代谢和免疫稳态的发展和维持至关重要,影响人体营养以及胃肠道功能和完整性[6,7]。越来越多的科学证据表明,微生物与宿主的相互作用不仅影响肠道环境,还影响远端器官[8,9]。有证据表明,肠道微生物群可以显著影响先天免疫系统和适应性免疫系统,并在促进或保护疾病发展方面发挥双重作用[10]。研究表明,肠道微生物群组成的紊乱可能与几种自身免疫性疾病有关,包括系统性红斑狼疮、类风湿性关节炎和多发性硬化症[11]。
在重症肌无力患者和动物模型中都观察到肠道微生物群组成的改变。一项病例对照研究表明,重症肌无力(MG)组与健康对照组之间的肠道微生物群多样性和丰度存在差异。具体来说,MG组表现出厚壁菌门、梭状芽胞杆菌、真杆菌和prausnitzii的水平降低。相反,MG组中变形菌门、拟杆菌门、链球菌和副菌门的水平较高。健康对照组的梭状芽孢杆菌水平约为MG组的3倍[12]。根据German Moris等人的研究,MG患者中Verrucomicrobiaceae和Bifidobacteriaceae的丰度低于健康对照组,而Bacteroidetes和Desulfovibrionaceae的丰度则高于健康对照组[1]。动物实验表明,将MG小鼠的微生物群移植到无菌小鼠身上会导致运动功能下降,而通过混合微生物群(由MG和健康微生物群组成)可以恢复运动功能[2]。这表明肠道微生物群可能在MG的发展中起作用。
然而,观察性研究主导了目前的大多数研究;虽然表明肠道微生物群与MG之间存在关联,但观察性研究得出的结论往往是基于“关联”而不是“因果关系”。此外,人口统计学、合并症、药物和饮食等混杂因素也不能排除。而且,在动物身上进行的实验并不能保证在人类身上得到同样的结果。孟德尔随机化方法旨在减轻这些限制。
孟德尔随机化是近年来广泛应用于流行病学因果关系推断的一种数据分析方法。传统的流行病学因果推断受到反向因果关系和混杂因素的阻碍。由于人类医学和试验设计的伦理限制,随机对照试验难以实施。MR通过使用基因变异作为工具变量(IVs)(通常是单核苷酸多态性(SNPs))来推断治疗暴露对结果的因果影响,从而弥补了这些局限性[13]。孟德尔遗传定律确保了配子形成过程中亲本等位基因随机分布给后代,从而最大限度地减少了出生后环境、社会经济地位和行为因素等常见混杂因素的干扰[13,14]。这种合理的因果顺序导致对真正因果效应的更准确估计。本研究旨在利用磁共振成像方法阐明肠道微生物群变化与MG之间的关系。
我们进行了双向磁共振分析,以调查肠道微生物群与MG之间的因果关系;该分析的流程图和设计如图1A所示。首先,肠道微生物群和MG的全基因组关联研究(GWAS)数据分别来自Mibiogen Consortium[15]和FinnGen Research Project。从GWAS数据中检索遗传变异并作为IVs使用。然后,使用R软件包“TwoSampleMR”(0.5.6)进行双样本MR,其中包括五种MR方法。进行敏感性分析,包括多效性[16]和异质性检验[17],以及遗漏分析。MR-PRESSO用于检测和校正异常值[18]。最后,进行反向分析,得出全面的因果关系结论。
图1
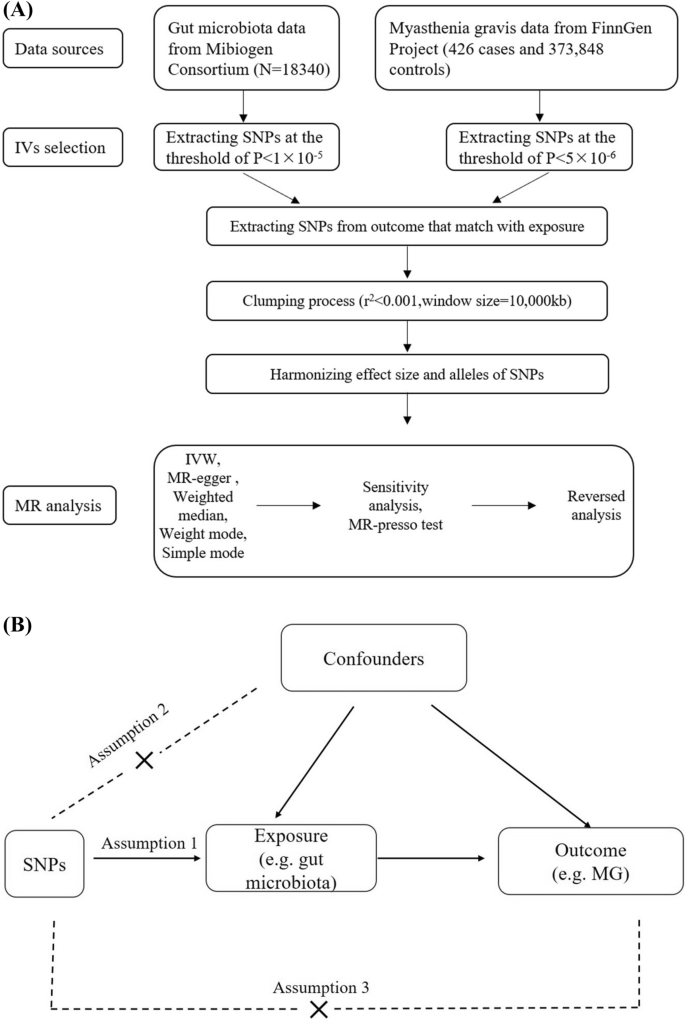
研究设计和孟德尔随机化核心假设。A双向孟德尔随机化的数据来源与研究设计。孟德尔随机化中的三个核心假设
对于偏差最小的结果,在使用MR方法时坚持三个关键假设是至关重要的:(1)IVs与暴露显著相关;(2) IVs不存在任何与暴露-结果相关的混杂因素,(3)IVs仅通过暴露影响结果[19](图1B)。
数据源
本研究中使用的肠道微生物群汇总数据来自全球联盟MiBioGen最近进行的全基因组关联研究[15]。该研究的庞大数据库收集了18340名主要来自欧洲的参与者(N=13266)的16S核糖体RNA基因测序和基因型数据。研究人员收集了211种细菌的特征,隶属于9门、16纲、20目、35科、131属。MG的数据收集自FinnGen研究项目,包括426例MG诊断病例和373848例欧洲对照。本研究中使用的数据来自公开可用的存储库,不需要任何进一步的伦理批准或患者同意。
在正向分析中,暴露变量为肠道微生物群,结果变量为MG。选择IVs的过程如图1A所示。首先,根据前人的研究[20],选取一组低于1 × 10?5的全基因组显著性阈值的snp作为IVs。其次,为了确保每个IV是独立的,我们利用欧洲1000基因组计划参考数据集将snp聚集在一起,r2值为0.001,聚集窗口为10,000 kb[21]。第三,我们协调SNP对结果和暴露的影响,确保它们指向相同的等位基因,纠正非回文SNP的链,并去除所有回文序列。f统计量计算了每个SNP作为工具变量的效力,其值超过10表示强大的工具。
在以MG作为暴露变量的反向因果关系研究中,一组低于全基因组统计显著性阈值(5 × 10?8)的snp被用作iv;然而,只有一个SNP达到了这个阈值。因此,选择第二个p值小于全基因组显著性水平(5 × 10?6)的snp来确定潜在的因果关系。剩余的流量和参数与前向MR相同。
为了评估肠道微生物群与MG之间的因果关系,采用了反方差加权(IVW)、MR-Egger回归、加权中位数、简单模型和加权模型等多种方法。初步分析采用IVW法,其他4种方法作为IVW法的补充[22]。IVW方法使用荟萃分析方法来结合每个SNP的Wald比,当所有IVW都有效时,它提供了最精确的估计[23]。MR-Egger回归依赖于“工具强度独立于直接效应”的假设,即暴露和结果是独立的。它的精度和统计能力较低,但可用于校正水平多效性[16]。当高达50%的信息来自无效的遗传变异时,加权中位数法提供了对因果效应的最无偏估计[24]。作为附加步骤,我们使用加权模式和简单模式来提高精度和稳定性[25]。
进行了各种敏感性分析来评估结果的强度。采用MR-Egger截距,根据回归截距线到零的距离来识别水平多效性[26]。科克伦Q检验是一种用于评估不同IVs之间异质性的方法。如果Cochran’s Q检验的p值小于预先定义的显著性水平(0.05),则认为存在显著异质性。反之,如果p值大于显著性水平(0.05),则认为IVs之间不存在显著异质性[27]。使用留一敏感性方法来评估一个SNP是否显著影响因果关系估计[28]。MR-PRESSO测试用于检测和去除可能的异常值,然后提供估计,从而校正水平多效性[18]。最后,使用公式β2/SE2计算每个SNP的f统计量[28],其中β和SE分别表示效应等位基因的估计误差和标准误差[29]。在本研究中,f统计量小于或等于10的SNP(定义为弱IVs)被排除在MR分析之外。为了调整多重比较,我们采用了Bonferroni校正,对肠道微生物群和MG给出了p=2.37 × 10?4(0.05/211)的截止值,校正后的p > 0.05被认为表明存在关联[30]。
所有统计分析均使用R 4.2.2版本进行。MR分析使用两样品MR(版本0.5.6)[28]和MR- presso(版本1.0)[31]R包进行。
摘要。
介绍
材料与方法
结果
讨论
限制
结论
数据和材料的可用性
缩写
参考文献。
致谢。
作者信息
道德声明
# # # # #
IVW分析结果显示,clostridiaceae科(OR=0.424,95%CI 0.202-0.889,p=0.023)、Defluviitaleaceae科(OR=0.537,95%CI 0.290-0.995,p=0.048)、Enterobacteriaceae科(OR=0.341,95%CI 0.135-0.865,p=0.023)和一个未知属(OR=0.407,95%CI 0.209-0.793,p=0.008)与MG的风险呈负相关(表1和图2A-D),而Lachnoclostridium属(OR=2.431,95%CI 1.047-5.647,p=0.05)与MG的风险呈负相关。p=0.039)与MG风险呈正相关(表1和图2E)。另外四种方法得到了相同的结果。
表1基因预测肠道微生物群与重症肌无力之间因果关系的MR研究结果
图2
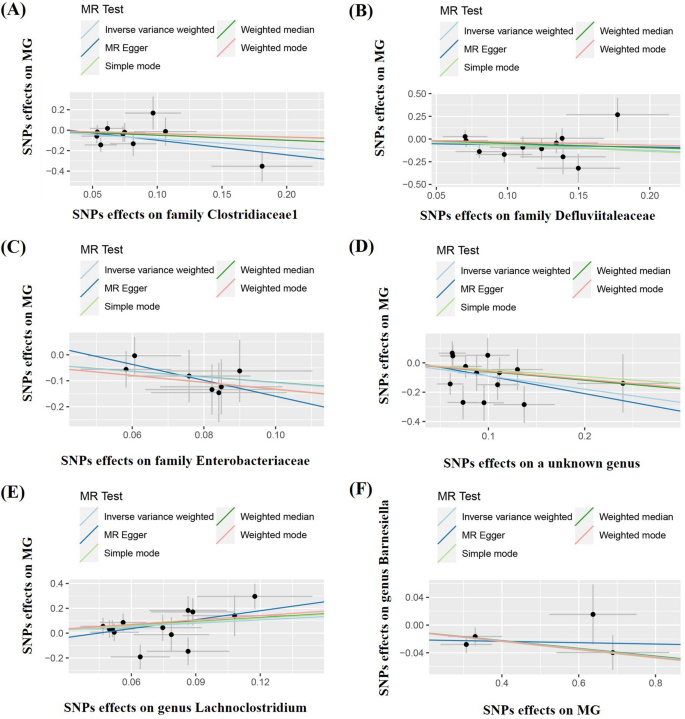
肠道菌群与MG之间因果关系的散点图
反向IVW分析显示,MG与Barnesiella属的丰度较低相关(OR=0.945,95%CI 0.906 ~ 0.985, p=0.008)。另外四种方法得到的结果相同(表2和图2F)。
表2基因预测重症肌无力与肠道菌群之间的MR研究结果
敏感性分析未发现潜在异质性或多效性(p > 0.05),见表1和表2。此外,在MR-PRESSO或留一分析中没有发现显著的异常值(表1和图3)。估计的f统计量均大于10,表明不存在弱IVs。关于最终snp和相应的漏斗图和森林图的更多细节汇总在附加文件1中。
图3
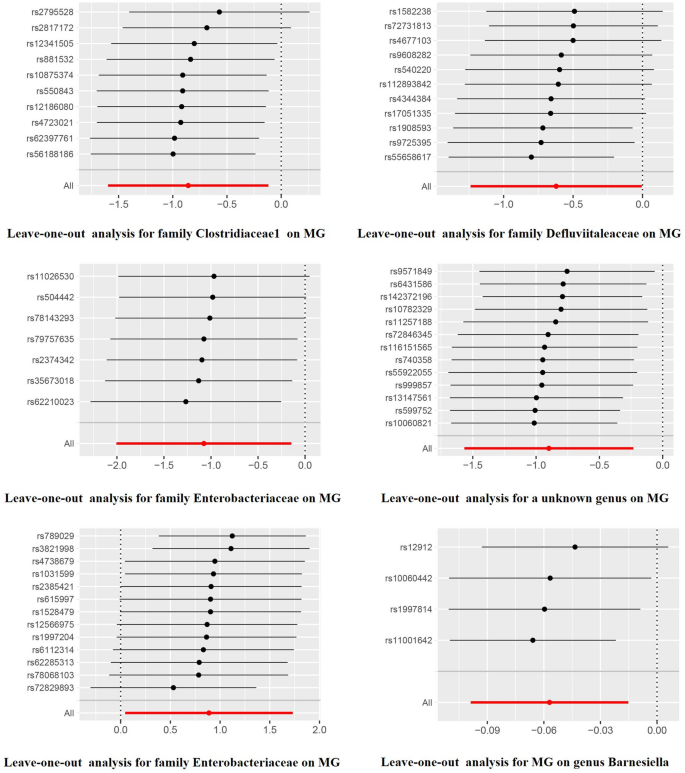
肠道微生物群和MG之间的留一分析
已经提出了几种假说来解释MG的发病机制;这些包括针对乙酰胆碱受体(AchR)的抗体,CD4 + T细胞在MG发病机制中的作用,以及CD4 + T细胞亚型和细胞因子在MG和实验性自身免疫性重症肌无力中的作用[32,33,34]。此外,在缺乏抗achr抗体的MG患者中也观察到其他自身抗体,如抗麝香抗体[35]。值得注意的是,人们越来越关注肠道菌群在疾病中的作用。单一微生物物种或全球共生群落的变化都可能通过改变致病性和保护性免疫反应之间的平衡,在自身免疫性疾病中发挥治疗作用[36]。在类风湿关节炎和炎症性肠病的病例中,这方面已经有所改善[37,38]。我们的研究强调了肠道微生物群变化对MG的影响,以及MG引起特定肠道微生物群水平变化的潜力。目前,这是第一个双样本双向磁共振研究,以证明肠道微生物群和MG之间的关系。其中Clostridiaceae1、Defluviitaleaceae、Enterobacteriaceae和一个未知属与MG呈负相关,Lachnoclostridium被认为是MG的潜在危险因素。此外,反向分析显示MG的存在与巴尼斯氏菌的水平呈负相关。
我们的研究表明,多种细菌在MG发病中起抑制作用,具体机制可能多种多样。
Foxp3 + CD4 + Treg细胞通过调节自身反应性T细胞的数量和抑制自身反应性B细胞的活性来调节致病抗体的产生,从而降低疾病的严重程度和进展,在维持自身耐受和免疫稳态中发挥关键作用[39]。MG患者外周血淋巴细胞Foxp3 + CD4 + Treg细胞的频率明显不足[12]。因此Foxp3 + CD4 + Treg细胞的丰度对于预防和治疗MG至关重要,成为目前MG发病机制研究的主要焦点。研究表明,肠道微生物群,特别是梭状芽胞杆菌,可以影响Foxp3 + CD4 + Treg细胞和表面T细胞受体(TCR)的数量。Foxp3 + CD4 + Treg细胞上的TCR能够识别共生菌亚群,诱导原生CD4 + T细胞分化为抗原特异性Foxp3 + CD4 + Treg细胞,并以此增加其数量。一个突出这一点的例子是梭状芽胞杆菌。梭状芽胞杆菌能够在靠近上皮的黏液层上定殖,增加2,3-双加氧酶和TGF-β1的表达[40,41]。这些变化可能促进未成熟T细胞的分化,导致Foxp3 + CD4 + Treg细胞的形成。这种生物学机制提供了一种保护作用,在无菌小鼠和人类结肠中已经观察到这种作用[42,43]。增殖激活受体(proliferator-activated receptor, PPARγ)参与调节免疫细胞的增殖和分化,可通过诱导分化增加Foxp3 + CD4 + Treg细胞的数量[44]。已证实链球菌通过抑制某些途径或免疫细胞功能激活PPARγ,从而调控PPARγ及其配体15d-PGJ2[45]。其治疗其他免疫性疾病如类风湿关节炎和炎症性肠病的疗效已得到证实[46,47]。这些发现强调了这样一个事实,肠道微生物群可以通过PPARγ等因子影响转录调节,从而导致免疫系统反应的紧密协调平衡。
短链脂肪酸(SCFAs)是肠道菌群产生的非营养性物质,具有重要的生理调节功能。它们提供人体所需的部分能量,保护肠黏膜屏障,抑制肠道炎症,调节免疫反应[48,49]。SCFAs是另一种可以调节Foxp3 + CD4 + Treg细胞的代谢物。它们对T细胞影响深远,直接调控T细胞向Foxp3 + CD4 + Treg细胞的分化[43,50]。因此,肠道菌群可能通过增加微生物代谢物(microbial metabolites, SCFAs)来增加Foxp3 + CD4 + Treg细胞的数量,间接发挥保护作用。
调节肠道菌群增加Foxp3 + CD4 + Treg细胞的数量可能成为治疗MG的新策略。利用益生菌调节MG的过程已经在动物模型中得到验证,在MG模型大鼠中,观察到五种益生菌菌株(嗜热链球菌、reuteri乳杆菌、两歧双歧杆菌、嗜酸乳杆菌和干酪乳杆菌)的混合物可以抑制促炎淋巴细胞反应,降低AchR抗体水平,这种效果也通过增加Foxp3 + CD4 + Treg细胞的数量来实现。然而,适用于人体的益生菌的具体菌株,以及对不同类型MG的最大功效所需的剂量,还需要进一步研究。我们的研究没有显示出上述保护性细菌的阳性结果,即Clostridium和Streptococcus,这可能是由于样本量小的原因。然而,本研究中获得的保护性细菌,如Clostridiaceae1、Defluviitaleaceae、Enterobacteriaceae以及一种未知类型的细菌,拓宽了未来研究的细菌范围。
在先前的动物实验中,已经证明将MG小鼠的肠道微生物群移植到无菌小鼠体内会导致运动障碍,但具体的细菌菌株仍未确定。这项研究首次证明了Lachnoclostridium是MG发病的一个危险因素。Lachnoclostridium是一种重要的产生SCFAs的细菌,在体内具有抗炎作用[51,52]。虽然先前的研究表明scfa对foxp3 + CD4 + treg有有益的影响,但Lachnoclostridium与MG的关联是矛盾的。造成这种现象的原因是复杂的。一方面,scfa并不总是具有神经保护作用。例如,在α -突触核蛋白过表达的小鼠模型中,口服SCFAs可加重运动障碍[53],这表明SCFAs可能在不同的病理背景下产生不同的作用。另一方面,Lachnoclostridium的16S rRNA基因测序是在属水平上进行的,无法确定是否与特定菌株或细菌种有关,这一结果不一致。此外,粪便代谢物目前被认为是肠道微生物群功能的外在表现[2]。除了上述SCFA外,研究还表明肠道微生物群与一系列代谢生物标志物(如缬氨酸、亮氨酸、黄嘌呤、胞嘧啶、萘和儿茶酚)之间存在显著相关性[2]。这些代谢产物可能支持肠道微生物群紊乱与MG相关的观点,因为肠道微生物群可能通过氨基酸、核苷酸和微生物代谢等代谢途径影响MG的发生[54,55]。这是先前提出的抗体介导的MG发病机制的潜在新补充。然而,应该承认的是,本研究中发现的Lachnoclostridium是否参与了这些代谢途径,以及它如何参与MG的发病机制尚不清楚,这一点值得进一步研究。
在反向MR分析中,发现MG导致巴尼斯氏菌的减少。先前的研究表明,MG可以改变肠道微生物群中细菌群的相对丰度。具体来说,它会导致梭状芽孢杆菌的减少[56]。相比之下,本研究仅发现MG与Barnesiella呈负相关,未观察到与Clostridium的相关性。一项病例对照研究发现,MG组的细菌多样性和丰度低于健康对照组[12]。因此,微生物多样性指数可作为评估MG严重程度的新的临床工具[2]。此外,MG患者肠道菌群的变化与一些临床参数略有关联。结合肠道微生物群和肠道代谢物可以帮助将MG受试者与健康对照组区分开来[2]。鉴于目前缺乏关于巴尼斯氏菌的详细数据,很难评估其水平变化在评估疾病方面的功效。然而,值得注意的是,这项研究的发现可能为未来的研究提供重要的见解。
我们的研究有几个限制需要考虑。首先,我们分析的样本人口只包括欧洲人。因此,研究结果在多大程度上可以推广到非欧洲人群尚不清楚。其次,由于数据集的限制,无法在属水平上探索肠道微生物群与MG之间的因果关系。第三,经过多次比较,我们在研究中发现的关联只是暗示了因果关系,而不是确定的。最后,我们使用的是汇总统计数据,而不是原始数据,这使得对MG进行亚组分析具有挑战性。因此,在未来需要更全面的数据来证实我们的发现。
我们的研究提供了肠道微生物群和MG之间双向因果关系的证据。我们确定了与MG相关的特定类型的微生物,这为了解该病的发病机制和制定治疗策略提供了新的窗口。然而,需要更多的基础和临床研究来阐明肠道微生物群在MG发病机制中的确切作用和治疗潜力。
本研究使用的原始数据,以及孟德尔随机化分析、异质性和多效性检验的结果。
ccDownload: /内容/ pdf / 10.1186 / s13578 - 023 - 01163 - 8. - pdf